





 und wie man sie reduziert")





Überblick
Marfan-Syndrom ist eine Erkrankung, die das Bindegewebe des Körpers betrifft und das Herz, die Aorta und andere Körperteile schädigt. Dieser komplexe Zustand erfordert einen spezialisierten und erfahrenen Behandlungsansatz.
-
Erfahren Sie mehr über das Marfan-Syndrom.
-
Laden Sie einen kostenlosen Leitfaden zur Erkrankung und Behandlung des Marfan-Syndroms herunter.
Veränderungen des Herzens und der Blutgefäße beim Marfan-Syndrom
Etwa 90 % der Menschen mit Marfan-Syndrom entwickeln Veränderungen in ihrem Herzen und ihren Blutgefäßen:
Blutgefäße
Die Wände der Blutgefäße, insbesondere die Aorta, die Hauptschlagader, die Blut vom Herzen zum Rest des Körpers transportiert, werden infolge des Marfan-Syndroms schwach und dehnen sich aus.
Dies erhöht das Risiko für Folgendes:
- Aortenaneurysma (eine Ausbuchtung, ähnlich einem Ballon),
- Aortendissektion (ein Reißen oder Trennen der Schichten der Aorta)
- Aortenruptur (Platzen), was zu einem medizinischen Notfall oder sogar zum Tod führen kann.
Herz
Die Herzklappen, insbesondere die Mitralklappe, können vom Marfan-Syndrom betroffen sein. Die Klappensegel werden aufgrund des Marfan-Syndroms schlaff und schließen nicht dicht, sodass Blut rückwärts über die Klappe austreten kann (Mitralklappenprolaps, auch MVP genannt).
Ein Mitralklappenprolaps erhöht die Belastung des Herzens und kann Kurzatmigkeit, übermäßige Müdigkeit oder Herzklopfen (Flattern in der Brust) verursachen. Der abnormale Fluss kann ein Herzgeräusch verursachen, das mit einem Stethoskop gehört werden kann. Im Laufe der Zeit kann sich das Herz vergrößern und es kann zu einer Herzinsuffizienz kommen.
Ein weiteres Problem, das beim Marfan-Syndrom beobachtet wird, ist die Dilatation (Verbreiterung) der Aortenwurzel, dem Bereich, in dem die Aorta auf die Aortenklappe trifft. Das Marfan-Syndrom kann dazu führen, dass die Aortenklappe gedehnt wird und undicht wird.
Arrhythmie (abnormaler Herzrhythmus) kann bei einigen Patienten mit Marfan-Syndrom auftreten. Dies hängt oft mit MVP zusammen.
Operation des Marfan-Syndroms
Die Operation des Marfan-Syndroms zielt darauf ab, eine Dissektion oder Ruptur zu verhindern und Klappenprobleme zu behandeln.
Aorta-Chirurgie
Etwa 40 % der Patienten mit Marfan-Syndrom sterben sofort, wenn eine Aortendissektion auftritt. Das Sterberisiko liegt zwischen 1 % und 3 % pro Stunde nach dem Dissektionsereignis. Selbst bei einer Notoperation liegt das Sterberisiko zwischen 10 % und 20 %. Das Ziel ist es, eine Operation vor der Dissektion durchzuführen, weil:
- Frühe Ergebnisse sind besser (mehr als 98 % Überlebensrate)
- Die langfristige Lebenserwartung ist besser.
Die normale Aorta misst etwa 2,54 Zentimeter. Wenn der Durchmesser der Aorta mehr als 4,7 Zentimeter beträgt oder wenn sich die Aorta schnell vergrößert, wird eine Operation empfohlen.
Vergleichen Sie das Aortenaneurysma links mit dem normalen Herzen rechts.

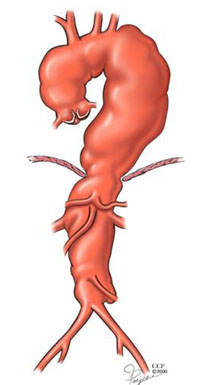
Alle Teile der Aorta können dilatieren oder dissezieren
Die Entscheidung für eine Operation basiert auf folgenden Faktoren:
- Größe der Aorta
- Erwartete normale Größe der Aorta
- Geschwindigkeit des Aortenwachstums
- Zeitalter
- Höhe
- Geschlecht
- Familiengeschichte der Aortendissektion
Bei einer nicht notfallmäßigen Aortenoperation beträgt die Überlebensrate mehr als 98 Prozent.
Bei einer Operation wird der dilatierte Teil der Aorta durch ein Transplantat ersetzt. Dies kann zwei Techniken umfassen:
- Traditionelle Methode: Ersetzen Sie die Aorta durch ein Transplantat und die Aortenklappe durch eine mechanische Klappe
- Ventilschonende Methode: Ersetzen Sie die Aorta durch ein Röhrchentransplantat und implantieren Sie die native Klappe erneut
Traditionelle Methode
Während der Operation entfernt der Herzchirurg den Dissektionsbereich oder das Aneurysma. Aufgrund der Bindegewebserkrankung neigt die Aorta zum Reißen, daher muss der Herzchirurg große Sorgfalt walten lassen.
Eine am Ende des Aortentransplantats angebrachte mechanische Klappe wird an den Ring (Öffnung) der Aortenklappe genäht.
Die Koronararterien werden durch kleine in das Transplantat geschnittene Knopflöcher wieder an dem Aortentransplantat befestigt. Dann wird das andere Ende des Dacron-Transplantats an das Gewebe der Aorta genäht.

Transplantat mit befestigtem mechanischem Ventil (mit freundlicher Genehmigung von St.Jude Medical™)
Valve sparing oder Valve Preserving (Reimplantationschirurgie)
Es gibt zwei Methoden, um die Aorta zu ersetzen, ohne die Aortenklappe zu ersetzen:
- Ventilerhaltende Reimplantationsmethode
- Ventilumbauverfahren.
Beide werden bei jungen Menschen angewendet, deren Aorten nicht zu stark vergrößert sind und die Aortenklappe nicht beschädigt ist.
Die klappenerhaltende Reimplantationsmethode wird für Patienten mit Marfan-Syndrom bevorzugt.
Nach einer Aortentransplantation verbleiben die Patienten in der Regel 5 bis 10 Tage im Krankenhaus. Nach einer Erholungsphase von sechs bis acht Wochen ist eine Rückkehr zu den Aktivitäten zu erwarten.
- Erfahren Sie mehr über die Valve-Sparing-Methode
Schritt-für-Schritt-Ansatz

1. Die Koronararterien zweigen von der Aorta ab. Die Aorta wird eröffnet und der Dissektionsbereich wird entfernt

2. Die Koronararterien werden von der erkrankten Aorta getrennt. Sie können die native Aortenklappe des Patienten sehen.

3. Das Dacron-Transplantat wird an den Ring der nativen Klappe genäht.

4. Die Koronararterien werden wieder am Transplantat befestigt. Das andere Ende des Transplantats wird an die Aorta genäht.

5. Die Operation ist abgeschlossen.
Reparatur oder Austausch von Ventilen
Eine undichte Aorten- oder Mitralklappe (Regurgitation), die zu Veränderungen im linken Ventrikel (linke untere Herzkammer, Hauptpumpkammer des Herzens) oder Herzinsuffizienz führt, erfordert eine Operation, um die Klappe zu reparieren oder zu ersetzen.
Dies kann mit traditionellen oder minimal-invasiven Techniken erfolgen.
- Erfahren Sie mehr über Klappenchirurgie.
Es wird empfohlen, dass sich Menschen mit Marfan-Syndrom einer Operation durch Herzchirurgen unterziehen, die in dieser Art von Operation erfahren sind. Diejenigen, die sich einer Operation unterziehen, benötigen immer noch lebenslange Nachsorge und vorbeugende Maßnahmen, um ein zukünftiges Fortschreiten der Krankheit zu verhindern. Die Mehrheit der Patienten, die vor der Dissektion operiert wurden, benötigen keine weiteren Operationen. Diejenigen, die nach der Dissektion operiert werden, sind jedoch einem größeren Risiko für zukünftige Operationen ausgesetzt, um andere Abschnitte der Aorta zu reparieren.
Ein besseres Verständnis des Marfan-Syndroms in Kombination mit früherer Erkennung, sorgfältiger Nachsorge und sichereren Operationstechniken haben zu besseren Ergebnissen für die Patienten geführt.
Ressourcen
zusätzliche Information
Von der Cleveland Clinic
-
Unser Aortenzentrum, spezialisiert auf die Behandlung des Marfan-Syndroms und Bindegewebserkrankungen sowie die Ärzte und Chirurgen, die Aortenaneurysmen behandeln
- Aortenerkrankung, Marfan-Syndrom und Bindegewebserkrankungen
- Aorta-Chirurgie
- Ihre Aorta
- Webchat-Transkripte für Aortenerkrankungen
- Videos über Aortenerkrankungen
- Ergebnisse
Andere Ressourcen
-
National Institute of Arthritis and Musculoskeletal and Skin Disease, Fragen und Antworten zum Marfan-Syndrom
- Nationale Marfan-Stiftung
- American Heart Association
: Was ist Amniozentese?")
")
Discussion about this post