











Was ist die Creutzfeldt-Jakob-Krankheit?
Die Creutzfeldt-Jakob-Krankheit ist eine degenerative Hirnstörung, die zu Demenz und letztendlich zum Tod führt. Die Symptome der Creutzfeldt-Jakob-Krankheit (CJD) können denen anderer demenzähnlicher Hirnstörungen wie Alzheimer ähneln. Die Creutzfeldt-Jakob-Krankheit verläuft jedoch in der Regel viel schneller.

CJD erregte in den 1990er Jahren die öffentliche Aufmerksamkeit, als einige Menschen im Vereinigten Königreich nach dem Verzehr von Fleisch von erkrankten Rindern eine Form der Krankheit entwickelten – Variante CJD (vCJD). Die „klassische“ Creutzfeldt-Jakob-Krankheit wurde jedoch nicht mit kontaminiertem Rindfleisch in Verbindung gebracht.
Obwohl schwerwiegend, ist CJD selten und vCJD ist die am wenigsten verbreitete Form. Weltweit wird jedes Jahr schätzungsweise ein Fall von CJD pro Million Menschen diagnostiziert, am häufigsten bei älteren Erwachsenen.
Symptome der Creutzfeldt-Jakob-Krankheit
Die Creutzfeldt-Jakob-Krankheit ist in der Regel innerhalb weniger Monate durch eine rasche geistige Verschlechterung gekennzeichnet. Erste Symptome sind typischerweise:
- Persönlichkeitsveränderungen
- Angst
- Depression
- Gedächtnisverlust
- Gestörtes Denken
- Verschwommenes Sehen oder Blindheit
- Schlaflosigkeit
- Schwierigkeiten beim Sprechen
- Schluckbeschwerden
- Plötzliche, ruckartige Bewegungen
Mit fortschreitender Krankheit verschlechtern sich die psychischen Symptome. Die meisten Menschen fallen schließlich ins Koma. Herzinsuffizienz, Ateminsuffizienz, Lungenentzündung oder andere Infektionen sind im Allgemeinen die Todesursache. Der Tod tritt normalerweise innerhalb eines Jahres ein.
Bei Menschen mit der selteneren vCJD können psychiatrische Symptome am Anfang stärker ausgeprägt sein, wobei sich Demenz – der Verlust der Fähigkeit zum Denken, Denken und Erinnern – später in der Krankheit entwickelt. Darüber hinaus betrifft diese Variante Menschen in einem jüngeren Alter als die klassische CJD und scheint eine etwas längere Dauer zu haben – 12 bis 14 Monate.
Ursachen der Creutzfeldt-Jakob-Krankheit
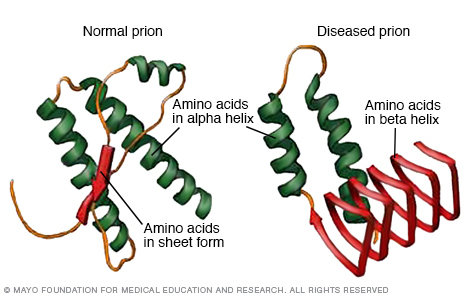
Die Creutzfeldt-Jakob-Krankheit und ihre Varianten gehören zu einer breiten Gruppe von menschlichen und tierischen Krankheiten, die als übertragbare spongiforme Enzephalopathien (TSE) bekannt sind. Der Name leitet sich von den schwammigen Löchern ab, die unter dem Mikroskop sichtbar sind und sich im betroffenen Gehirngewebe entwickeln.
Die Ursache der Creutzfeldt-Jakob-Krankheit und anderer TSEs scheinen abnormale Versionen einer Art Protein zu sein, das als Prion bezeichnet wird. Normalerweise sind diese Proteine harmlos. Aber wenn sie missgebildet sind, werden sie ansteckend und können normale biologische Prozesse schädigen.
Wie CJD übertragen wird
Das Risiko für CJD ist gering. Die Krankheit kann nicht durch Husten oder Niesen, Berühren oder sexuellen Kontakt übertragen werden. Die drei Arten, wie es sich entwickelt, sind:
- Sporadisch. Die meisten Menschen mit klassischer CJD entwickeln die Krankheit ohne ersichtlichen Grund. Dieser Typ wird als spontane CJD oder sporadische CJD bezeichnet und ist für die Mehrzahl der Fälle verantwortlich.
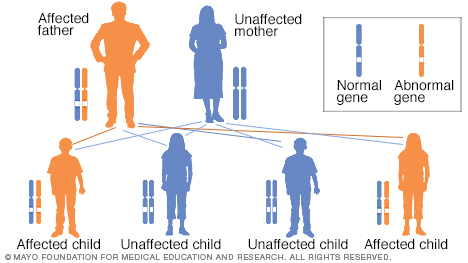
- Durch Vererbung. Weniger als 15 Prozent der Menschen mit CJD haben eine Familienanamnese der Krankheit oder testen positiv auf eine mit CJD assoziierte genetische Mutation. Dieser Typ wird als familiäre CJD bezeichnet.
-
Durch Kontamination. Eine kleine Anzahl von Menschen hat CJD entwickelt, nachdem sie während eines medizinischen Eingriffs wie einer Hornhaut- oder Hauttransplantation infiziertem menschlichem Gewebe ausgesetzt waren. Da Standardsterilisationsmethoden keine abnormalen Prionen zerstören, haben einige Menschen nach einer Gehirnoperation mit kontaminierten Instrumenten eine CJD entwickelt.
Fälle von CJD im Zusammenhang mit medizinischen Verfahren werden als iatrogene CJD bezeichnet. Die Variante CJD ist hauptsächlich mit dem Verzehr von Rindfleisch verbunden, das mit einer Rinderwahnsinnskrankheit (bovine spongiforme Enzephalopathie oder BSE) infiziert ist.
Risikofaktoren
Die meisten Fälle der Creutzfeldt-Jakob-Krankheit treten aus unbekannten Gründen auf, und es können keine Risikofaktoren identifiziert werden. Einige Faktoren scheinen jedoch mit verschiedenen Arten von CJD verbunden zu sein.
- Alter. Sporadische CJD entwickeln sich tendenziell später im Leben, normalerweise um das 60. Lebensjahr. Der Beginn der familiären CJD tritt etwas früher auf und vCJD hat Menschen in einem viel jüngeren Alter betroffen, normalerweise Ende 20.
-
Genetik. Menschen mit familiärer CJD haben eine genetische Mutation, die die Krankheit verursacht. Die Krankheit wird autosomal-dominant vererbt, was bedeutet, dass Sie nur eine Kopie des mutierten Gens von beiden Elternteilen erben müssen, um die Krankheit zu entwickeln. Wenn Sie die Mutation haben, beträgt die Chance, sie an Ihre Kinder weiterzugeben, 50 Prozent.
Genetische Analysen bei Menschen mit iatrogener und vCJD legen nahe, dass das Erben identischer Kopien bestimmter Varianten des Prion-Gens das Risiko für die Entwicklung von CJD erhöhen kann, wenn Sie kontaminiertem Gewebe ausgesetzt sind.
-
Exposition gegenüber kontaminiertem Gewebe. Menschen, die menschliches Wachstumshormon aus menschlichen Hypophysen erhalten haben oder Gewebetransplantate hatten, die das Gehirn bedecken (Dura Mater), sind möglicherweise einem Risiko für iatrogene CJD ausgesetzt.
Das Risiko einer vCJD-Infektion durch den Verzehr von kontaminiertem Rindfleisch ist schwer zu bestimmen. Wenn Länder Maßnahmen im Bereich der öffentlichen Gesundheit wirksam umsetzen, besteht im Allgemeinen praktisch kein Risiko.
Komplikationen durch Creutzfeldt-Jakob-Krankheit
Wie bei anderen Ursachen von Demenz betrifft die Creutzfeldt-Jakob-Krankheit sowohl das Gehirn als auch den Körper stark, obwohl CJD und seine Varianten normalerweise viel schneller fortschreiten. Menschen mit CJD ziehen sich normalerweise von Freunden und Familie zurück und verlieren schließlich die Fähigkeit, sie zu erkennen oder mit ihnen in Beziehung zu treten. Sie verlieren auch die Fähigkeit, für sich selbst zu sorgen, und viele fallen schließlich ins Koma. Diese Krankheit ist letztendlich tödlich.
Prävention der Creutzfeldt-Jakob-Krankheit
Es gibt keine Möglichkeit, sporadische CJD zu verhindern. Wenn Sie in der Familienanamnese an neurologischen Erkrankungen leiden, können Sie von einem Gespräch mit einem Genetikberater profitieren, der Ihnen dabei helfen kann, die mit Ihrer Situation verbundenen Risiken zu klären.
Vorbeugung von iatrogener CJD
Krankenhäuser und andere medizinische Einrichtungen befolgen explizite Richtlinien, um iatrogene CJD zu verhindern. Diese Maßnahmen umfassten:
- Ausschließliche Verwendung von synthetischem menschlichem Wachstumshormon anstelle der aus menschlichen Hypophysen stammenden Art
- Zerstörung von chirurgischen Instrumenten im Gehirn oder Nervengewebe von Personen mit bekannter oder vermuteter CJD
- Einweg-Kits für Wirbelsäulenhähne (Lumbalpunktionen)
Um die Sicherheit der Blutversorgung zu gewährleisten, sind Personen mit einem Risiko einer Exposition gegenüber CJD oder vCJD nicht berechtigt, Blut zu spenden.
VCJD verhindern
Das Risiko, an vCJD zu erkranken, ist äußerst gering. Eine sehr kleine Anzahl von vCJD-Fällen wurde weltweit gemeldet.
Regulierung potenzieller vCJD-Quellen
Die meisten Länder haben Maßnahmen ergriffen, um zu verhindern, dass mit BSE infiziertes Gewebe in die Lebensmittelversorgung gelangt, darunter:
- Strenge Einfuhrbeschränkungen für Rinder aus Ländern, in denen BSE verbreitet ist
- Beschränkungen für Tierfutter
- Strenge Verfahren für den Umgang mit kranken Tieren
- Überwachungs- und Testmethoden zur Verfolgung der Gesundheit von Rindern
- Einschränkungen, welche Teile von Rindern für Lebensmittel verarbeitet werden dürfen
Diagnose
Nur eine Gehirnbiopsie oder eine Untersuchung des Gehirngewebes nach dem Tod (Autopsie) kann das Vorhandensein der Creutzfeldt-Jakob-Krankheit bestätigen. Ärzte können jedoch häufig eine genaue Diagnose auf der Grundlage Ihrer medizinischen und persönlichen Vorgeschichte, einer neurologischen Untersuchung und bestimmter diagnostischer Tests stellen.
Die Untersuchung zeigt wahrscheinlich charakteristische Symptome wie Muskelzuckungen und Krämpfe, abnormale Reflexe und Koordinationsprobleme. Menschen mit CJD können auch Bereiche der Blindheit und Veränderungen in der visuell-räumlichen Wahrnehmung haben.
Darüber hinaus verwenden Ärzte diese Tests häufig, um CJD zu erkennen:
- Elektroenzephalogramm (EEG). Mit Hilfe von Elektroden auf Ihrer Kopfhaut misst dieser Test die elektrische Aktivität Ihres Gehirns. Menschen mit CJD und vCJD zeigen ein charakteristisch abnormales Muster.
- MRT. Diese Bildgebungstechnik verwendet Radiowellen und ein Magnetfeld, um Querschnittsbilder Ihres Kopfes und Körpers zu erstellen. Es ist besonders nützlich bei der Diagnose von Hirnstörungen, da es hochauflösende Bilder der weißen und grauen Substanz des Gehirns enthält.
- Wirbelsäulentests. Die zerebrale Wirbelsäulenflüssigkeit umgibt und polstert Ihr Gehirn und Ihr Rückenmark. In einem Test, der als Lumbalpunktion bezeichnet wird und im Volksmund als Wirbelsäulenhahn bezeichnet wird, verwenden Ärzte eine Nadel, um eine kleine Menge dieser Flüssigkeit zum Testen abzuziehen. Das Vorhandensein eines bestimmten Proteins in der Rückenmarksflüssigkeit ist häufig ein Hinweis auf CJD oder vCJD.
Behandlung der Creutzfeldt-Jakob-Krankheit
Es gibt keine wirksame Behandlung für die Creutzfeldt-Jakob-Krankheit oder eine ihrer Varianten. Eine Reihe von Medikamenten wurde getestet und hat keine Vorteile gezeigt. Aus diesem Grund konzentrieren sich Ärzte darauf, Schmerzen und andere Symptome zu lindern und Menschen mit diesen Krankheiten so angenehm wie möglich zu machen.
Zum Arzt gehen
Wenn Sie einen Termin bei einem Arzt vereinbaren, werden Sie möglicherweise sofort an einen Gehirnspezialisten (Neurologen) überwiesen.
Hier finden Sie einige Informationen, die Ihnen bei der Vorbereitung Ihres Termins helfen sollen.
Was du tun kannst
- Listen Sie Ihre Symptome auf, einschließlich aller, die möglicherweise nicht mit dem Grund zusammenhängen, aus dem Sie den Termin geplant haben.
- Notieren Sie wichtige persönliche Informationen, einschließlich der jüngsten Veränderungen im Leben.
- Medikamente auflisten, Vitamine und Nahrungsergänzungsmittel, die Sie einnehmen.
- Bringen Sie ein Familienmitglied oder einen Freund mit, wenn möglich. Jemand, der Sie begleitet, kann Ihnen helfen, sich an etwas zu erinnern, das Sie verpasst oder vergessen haben.
- Schreiben Sie eine Liste mit Fragen auf um Ihren Arzt zu fragen.
Bei der Creutzfeldt-Jakob-Krankheit sollten Sie Ihrem Arzt einige grundlegende Fragen stellen:
- Was verursacht wahrscheinlich meine Symptome?
- Was sind außer der wahrscheinlichsten Ursache andere mögliche Ursachen für meine Symptome?
- Welche Tests brauche ich?
- Was ist die beste Vorgehensweise?
- Gibt es Einschränkungen, denen ich folgen muss?
- Soll ich einen Spezialisten aufsuchen?
- Ich habe andere Krankheiten. Wie verwalte ich sie zusammen?
- Gibt es Broschüren oder andere Drucksachen, die ich haben kann? Welche Websites empfehlen Sie?
Zögern Sie nicht, andere Fragen zu stellen.
Was Ihr Arzt fragen kann
Zu den Fragen, die Ihr Arzt wahrscheinlich stellt, gehören:
- Wann haben Ihre Symptome begonnen?
- Waren Ihre Symptome kontinuierlich oder gelegentlich?
- Wie schwer sind Ihre Symptome?
- Was scheint Ihre Symptome zu verbessern, wenn überhaupt?
- Was, wenn überhaupt, scheint Ihre Symptome zu verschlimmern?
- Hat jemand in Ihrer Familie die Creutzfeldt-Jakob-Krankheit gehabt?
.
Discussion about this post